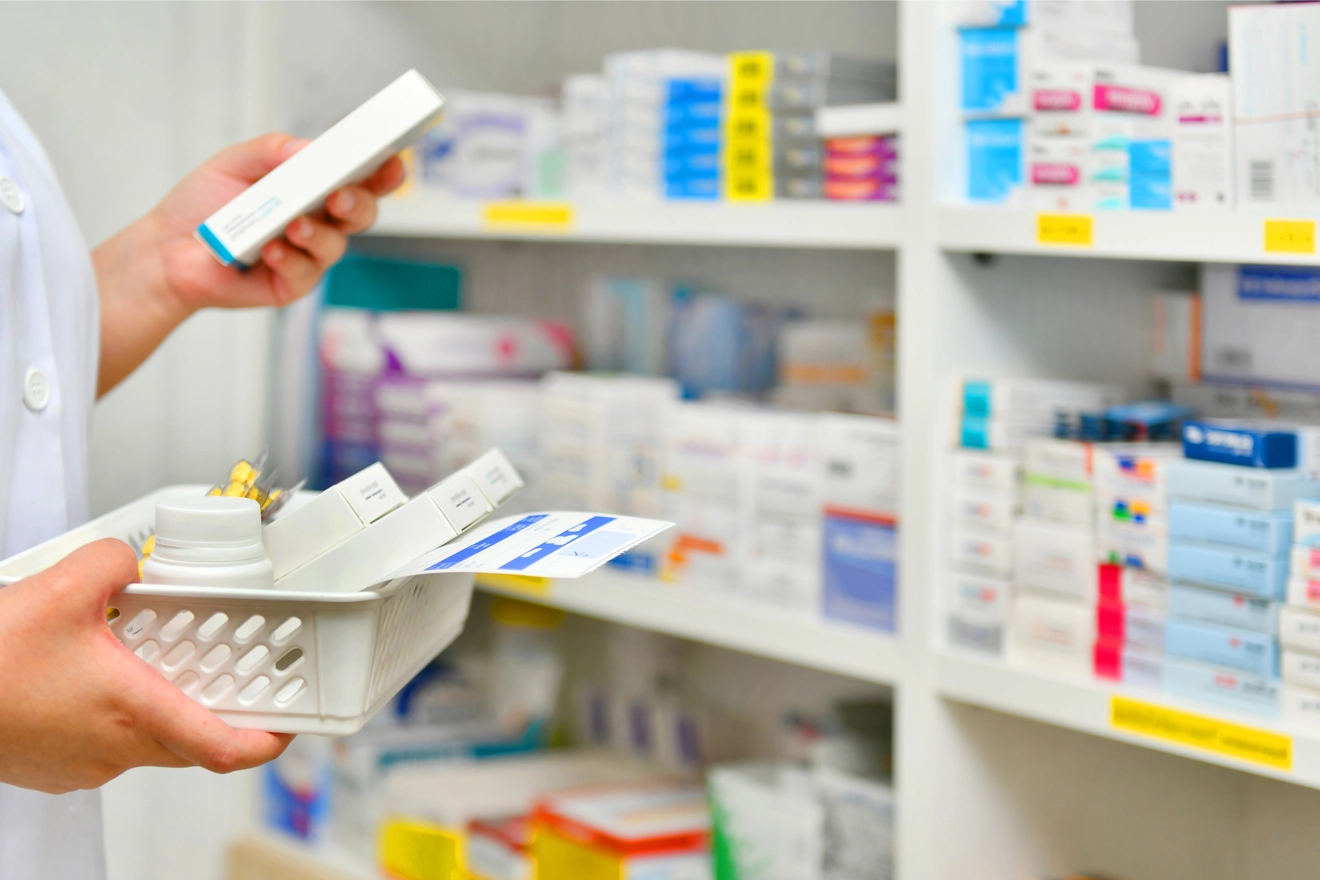
Introduction to Generics
Global Regulatory services for generic drug products have evolved over the past few decades. Owing to increasing demand for these products, given the lower prices offered by generic drug manufacturers, generic drug applications are being prioritized by Health Authorities (HAs) in the United States, Europe, and Canada. However, there exist a few untapped markets for generic drug development. Therefore, different Regulatory authorities like the US Food and Drug Administration (FDA), Health Canada, and the European Medicines Agency (EMA)/Medicines and Healthcare Products Regulatory Agency (MHRA) are supporting generic drug manufacturers to explore various opportunities related to generic drug application, dossier submissions, and generic drug registration to overcome the existing challenges during generic drug development.
Know More
Centralizing Regulatory Services for Generic Drugs
Regulatory requirements for generic drugs differ from one country to another. Therefore, to stay compliant and updated with the regional and global Regulatory standards, it is essential to understand the generic drug approval process and related Regulatory requirements for each country before submitting the MAA to the respective HAs. Freyr’s Regulatory team has an in-depth understanding of the local and regional generic drug application and dossier submission requirements across one hundred and twenty (120) countries.
What are Your Challenges?
Explore how Freyr can assist you in your global generic drug applications.



End-to-end Regulatory Services
Freyr offers comprehensive end-to-end global Regulatory services for generic drugs, which start from product development to lifecycle management and commercialization, to help companies maximize their asset value. Freyr’s global pool of Regulatory experts enables Life Sciences, Consumer Pharma, and Biomed companies to comprehend and manage the challenging and diverse Regulatory requirements to expand their business potential in new and existing markets.
Know MoreService Portfolios

What We Do
Freyr provides Regulatory solutions and services to large, mid & small global life sciences companies. Our highly-skilled Regulatory experts understand customer requirements and customize the process for end-to-end drug approvals & Regulatory Chemistry and Manufacturing and Controls (CMC) support through the optimum utilization of resources.
With an integrated drug delivery approach and efficient Regulatory roadmap strategy, we help our customers stay 100% compliant with the dynamic Regulatory requirements and thereby reduce the generic drug approval process timelines.
Drug Approval Process in the EU and US
The development of innovator drug products involves a huge investment of money and time, unlike generics, which are cost-effective and require less time to bring drug products to the market. Innovator drug manufacturers are required to submit the Investigational New Drug application (IND) in the USA, (IMPD application in Europe) and new drug application in the USA, (MAA in Europe) after the successful completion of clinical trials. Similarly, generic drug manufacturers are required to submit a generic drug application known as the Abbreviated New Drug Application (ANDA) in the USA and a Marketing Authorization Application (MAA) in Europe, post-completion of the bioequivalence studies.

Clinical Trails ~ 6 Years



Discovery Phase & PreClinical Studies 6 Years


Phase 1

Phase 2

Phase 3
Regulatory Review
0.5 - 2 Years
PMS

Exclusivity
- IND Submission to USFDA
IMPD Submission in EU - NDA Submission to USFDA
MAA Submission in EU - 12Years Market Exclusivity
8+2+1 Market Exclusivity


Product Development
~10-12 Months

Biobatch Production
and Stability data
~7-9 Months
Bioequivalence
Studies
~4-6 MonthsRegulatory Review
8-10 MonthsPMS
Exclusivity
- ANDA Submission to USFDA
MAA Submission in EU - 180 day Generic drug
Exclusivity Provided by USFDA
EU-No Exclusivity for Generics
Freyr Industry Leading Scale

150+
MAA

350+
DMFs

250+
ANDA Submissions

1500+
Annual Reports

4K+
Post-approval Activities

3K+
Renewals